Overview
Sickle-cell disease (SCD) [generally SICKLE- CELL ANEMIA (SCA)]is a genetic (blood) disorder which has no exact cure in medical sciences.
Although, this disease can be found at any place, in any community but this is more likely to be found in some specific communities spread across some particular places of the world. It is inherited in a child by its parent’s genes. It is difficult to say that when this disease was born and from where. But, it is noticed that this disease is more likely to b found in that places of the world where Malaria exists in large scale.
It is the assumption of scientists that to protect itself from the malarial parasite, the red blood cells may have changed form (mutation).
Sickle-cell disease (SCD), or sickle-cell anemia (SCA) or sometimes drepanocytosis, is a hereditary blood disorder, characterized by an abnormality in the oxygen-carrying hemoglobin molecule in red blood cells. This leads to a propensity for the cells to assume an abnormal, rigid, sickle-like shape under certain circumstances. Sickle-cell disease is associated with a number of acute and chronic health problems, such as severe infections, attacks of severe pain ("sickle-cell crisis"), and stroke, and there is an increased risk of death. Sickle-cell disease occurs when a person inherits two abnormal copies of the hemoglobin gene, one from each parent. Several subtypes exist, depending on the exact mutation in each hemoglobin gene. A person with a single abnormal copy does not experience symptoms and is said to have sickle-cell trait.
A child having the disease (by birth) always suffers from various problems like fever, cold, stomach ache, anemia after an age of six months.
There is a chance of damage to crucial organs like spleen, liver, lungs, kidneys etc. It may also lead to multiple organ failure.
ØPrognosis
Six percent of children suffering from Sickle – cell anemia could not survive after six months. A large mass survives only till an age of twenty – twenty one years. Those who survive further also have a shorter life expectancy and suffer from various problems in life.About 90% of patients survive to age 20, and close to 50% survive beyond the fifth decade. In 2001, according to one study performed in Jamaica, the estimated mean survival for sickle-cell patients was 53 years old for men and 58 years old for women with homozygous SCD.
The complications of sickle-cell disease can be prevented to a large extent with vaccination, preventative antibiotics, blood transfusion, and the drug hydroxyurea / hydroxycarbamide. A small proportion requires a transplant of bone marrow cells.
Almost 300,000 children are born with a form of sickle-cell disease every year, mostly in sub-Saharan Africa, but also in other countries such as the West Indies and in people of African origin elsewhere in the world. The condition was first described in the medical literature by the American physician James B. Herrick in 1910, and in the 1940s and 1950s contributions by Nobel prize-winner Linus Pauling made it the first disease where the exact genetic and molecular defect was elucidated.
WHAT IS SICKLE CELL ANEMIA?
In general, Sickle- cell anemia is a kind of secondary THALASSEMIA.
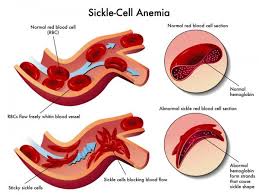
It is the most common form of sickle cell disease. SCD is a serious disorder in which the body makes sickle-shaped red blood cells. “Sickle-shaped” means that the red blood cells are shaped like a crescent moon.

Normal red blood cells are disc-shaped and look like doughnuts without holes in the center. They move easily through your blood vessels. Red blood cells contain an iron-rich protein called hemoglobin. This protein carries oxygen from the lungs to the rest of the body.
Sickle cell disease is an inherited blood disorder that affects red blood cells. People with sickle cell disease have red blood cells that contain mostly hemoglobin* S, an abnormal type of hemoglobin. Sometimes these red blood cells become sickle-shaped (crescent shaped) and have difficulty passing through small blood vessels.
Sickle cells are stiff and sticky. They tend to block blood flow in the blood vessels of the limbs and organs. Blocked blood flow can cause pain and organ damage. It can also raise the risk for infection.
- Other names for sickle- cell anemia:
- HbS disease
- Hemoglobin S disease
- Hemoglobin SS disease
- Sickle cell disease (a broad term that includes sickle cell anemia)
- Sickling disorder due to hemoglobin S.
HEMOGLOBIN is the main substance of the red blood cell. It helps red blood cells carry oxygen from the air in our lungs to all parts of the body. Normal red blood cells contain hemoglobin A. Hemoglobin S and hemoglobin C are abnormal types of hemoglobin. Normal red blood cells are soft and round and can squeeze through tiny blood tubes (vessels). Normally, red blood cells live for about 120 days before new ones replace them.
People with sickle cell conditions make a different form of hemoglobin A called hemoglobin S (S stands for sickle). Red blood cells containing mostly hemoglobin S do not live as long as normal red blood cells (normally about 16 days). They also become stiff, distorted in shape and have difficulty passing through the body's small blood vessels. When sickle-shaped cells block small blood vessels, less blood can reach that part of the body. Tissue that does not receive a normal blood flow eventually becomes damaged. This is what causes the complications of sickle cell disease.
§ Types of Sickle Cell Disease
There are several types of sickle cell disease. The most common are: Sickle Cell Anemia (SS), Sickle-Hemoglobin C Disease (SC)
Sickle Beta-Plus Thalassemia and Sickle Beta-Zero Thalassemia.
- Dependency factors of Sickle Cell Anemia
There are several factors, but mainly the disease depends upon variations in hemoglobin.
- Hemoglobin A (α2β2) (PDB 1BZ0) – The most common with a normal amount over 95%
- Hemoglobin A2 (α2δ2) – δ chain synthesis begins late in the third trimester and, in adults, it has a normal range of 1.5–3.5%
- Hemoglobin F (α2γ2) – In adults Hemoglobin F is restricted to a limited population of red cells called F-cells. However, the level of Hb F can be elevated in persons with sickle-cell disease and beta-thalassemia.
- Hemoglobin S (α2βS2) – A variant form of hemoglobin found in people with sickle cell disease. There is a variation in the β-chain gene, causing a change in the properties of hemoglobin, which results in sickling of red blood cells.
- Relation between SCD and Thalassemia
Thalassemia (British English: thalassaemia) is a form of inherited autosomal recessive blood disorders characterized by abnormal formation of hemoglobin. The abnormal haemoglobin formed results in improper oxygen transport and destruction of red blood cells. Thalassemia is caused by variant or missing genes that affect how the body makes hemoglobin, the protein in red blood cells that carries oxygen. People with thalassemia make less hemoglobin and have fewer circulating red blood cells than normal, which results in mild or severe anemia. Thalassemia will be present as microcytic anemia.
Thalassemia can cause significant complications, including iron overload, bone deformities, and cardiovascular illness. However, this same inherited disease of red blood cells may confer a degree of protection against malaria (specifically, malaria caused by the protozoan parasite Plasmodium falciparum), which is or was prevalent in the regions where the trait is common. This selective survival advantage of carriers (known as heterozygous advantage) may be responsible for perpetuating the mutation in populations. In that respect, the various thalassemias resemble another genetic disorder affecting hemoglobin, sickle-cell disease.
CAUSES
Sickle cell anemia is an inherited disease. People who have the disease two genes for sickle hemoglobin – one from each parent.
- GENETICS
Normally, humans have haemoglobin A, which consists of two alpha and two beta chains, haemoglobin A2, which consists of two alpha and two delta chains, and haemoglobin F, consisting of two alpha and two gamma chains in their bodies. Of these, haemoglobin F dominates until about 6 weeks of age then A dominates throughout life.
- Types of Sickle- Cell patients
- Sickle Carrier (AS):
These are also known as HETEROZYGOTES (carrying trait). They have one gene of sickle and the other one as normal. They have no symptoms of disease. They live a normal and healthy life. Some may have medical problems. They didn’t even know that they are carrying an infected gene. They play an important role in spread of the disease because their future generation is also affected.
- Sickle patient (SS):
These are also known as HOMOZYGOTES. They contain both the infected genes. They have all the symptoms of disease. In people heterozygous for HgbS (carriers of sickling haemoglobin), the polymerisation problems are minor, because the normal allele is able to produce over 50% of the haemoglobin. In people homozygous for HgbS, the presence of long-chain polymers of HbS distort the shape of the red blood cell from a smoothdoughnut-like shape to ragged and full of spikes, making it fragile and susceptible to breaking within capillaries. Carriers have symptoms only if they are deprived of oxygen (for example, while climbing a mountain) or while severely dehydrated. The sickle-cell disease occurs when the sixth amino acid, glutamic acid, is replaced by valine to change its structure and function; as such, sickle-cell anemia is also known as E6V. Valine is hydrophobic, causing the haemoglobin to collapse on itself occasionally. The structure is not changed otherwise. When enough haemoglobin collapses on itself the red blood cells become sickle-shaped.
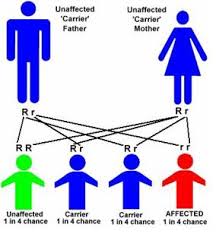
- SICKLE CELL TRAIT
People who inherit a sickle hemoglobin gene from one parent and a normal gene from the other parent have sickle cell trait. Their bodies make both sickle hemoglobin and normal hemoglobin.
People who have sickle cell trait usually have few, if any, symptoms and lead normal lives. However, some people may have medical complications.
People who have sickle cell trait can pass the sickle hemoglobin gene to their children. The following image shows an example of an inheritance pattern for sickle cell trait.
Example of an Inheritance Pattern for Sickle Cell Trait
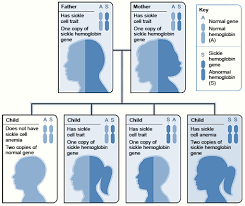
The image shows how sickle hemoglobin genes are inherited. A person inherits two hemoglobin genes—one from each parent. A normal gene will make normal hemoglobin (A). A sickle hemoglobin gene will make abnormal hemoglobin (S).
When both parents have a normal gene and an abnormal gene, each child has a 25 percent chance of inheriting two normal genes; a 50 percent chance of inheriting one normal gene and one abnormal gene; and a 25 percent chance of inheriting two abnormal genes.
HISTORY
The first modern report of sickle-cell disease may have been in 1846, where the autopsy of an executed runaway slave was discussed; the key finding was the absence of the spleen. There were also reports amongst African slaves in the United States exhibiting resistance to malaria but being prone to leg ulcers. The abnormal characteristics of the red blood cells, which later lent their name to the condition, was first described by Ernest Edward Irons (1877–1959), intern to the Chicago cardiologist and professor of medicine James B. Herrick (1861–1954), in 1910. Irons saw "peculiar elongated and sickle-shaped" cells in the blood of a man named Walter Clement Noel, a 20-year-old first-year dental student from Grenada. Noel had been admitted to the Chicago Presbyterian Hospital in December 1904 suffering from anemia. Noel was readmitted several times over the next three years for "muscular rheumatism" and "bilious attacks" but completed his studies and returned to the capital of Grenada (St. George's) to practice dentistry. He died of pneumonia in 1916 and is buried in the Catholic cemetery at Sauteurs in the north of Grenada. Shortly after the report by Herrick, another case appeared in the Virginia Medical Semi-Monthly with the same title, "Peculiar Elongated and Sickle-Shaped Red Blood Corpuscles in a Case of Severe Anemia." This article is based on a patient admitted to the University of Virginia Hospital on November 15, 1910. In the later description by Verne Mason in 1922, the name "sickle cell anemia" is first used. Childhood problems related to sickle cells disease were not reported until the 1930s, despite the fact that this cannot have been uncommon in African-American populations.
The Memphis physician Lemuel Diggs, a prolific researcher into sickle cell disease, first introduced the distinction between sickle cell disease and trait in 1933, although it took until 1949 until the genetic characteristics were elucidated by James V. Neel and E.A. Beet. 1949 was the year when Linus Pauling described the unusual chemical behaviour of hemoglobin S, and attributed this to an abnormality in the molecule itself. The actual molecular change in HbS was described in the late 1950s BY Vernon Ingram. The late 1940s and early 1950s saw further understanding in the link between malaria and sickle cell disease. In 1954, the introduction of hemoglobin electrophoresis allowed the discovery of particular subtypes, such as HbSC disease.
Large scale natural history studies and further intervention studies were introduced in the 1970s and 1980s, leading to the more widespread use of prophylaxis against pneumococcal infections amongst other interventions. The 1990s saw the development of hydroxyl carbamide, and reports of cure through bone marrow transplantation appeared in 2007.
EPIDEMIOLOGY
The highest frequency of sickle cell disease is found in tropical regions, particularly sub-Saharan Africa, tribal regions of India and the Middle-East. Migration of substantial populations from these high prevalence areas to low prevalence countries in Europe has dramatically increased in recent decades and in some European countries sickle-cell disease has now overtaken more familiar genetic conditions such as haemophilia and cystic fibrosis.In 2010, there were about 29,000 deaths attributed to sickle-cell disease globally.
Sickle-cell disease occurs more commonly among people whose ancestors lived in tropical and sub-tropical sub-Saharan regions where malaria is or was common. Where malaria is common, carrying a single sickle-cell allele (trait) confers aselective advantage—in other words, being a heterozygote is advantageous. Specifically, humans with one of the two allelesof sickle-cell disease show less severe symptoms when infected with malaria.
Africa
Three quarters of sickle-cell cases occur in Africa. A recent WHO report estimated that around 2% of newborns in Nigeria were affected by sickle cell anaemia, giving a total of 150,000 affected children born every year in Nigeria alone. The carrier frequency ranges between 10% and 40% across equatorial Africa, decreasing to 1–2% on the north African coast and <1% in South Africa. There have been studies in Africa that show a significant decrease in infant mortality rate, ages 2–16 months, because of the sickle-cell trait. This happened in areas that were known to be predominant areas of malarial cases.
United States
The prevalence of the disease in the United States is approximately 1 in 5,000, mostly affecting Americans of Sub-Saharan African descent, according to the National Institutes of Health. In the United States, about 1 out of 500 African-American children and 1 in every 36,000 Hispanic-American children born will have sickle-cell anaemia. It is estimated that sickle-cell disease affects 90,000 Americans. Most infants with SCD born in the United States are now identified by routine neonatal screening. Forty-four states along with the District of Columbia, Puerto Rico and the Virgin Islands currently provide universal neonatal screening for SCD. Sickle cell trait occurs among about 1:12 African-Americans and 1:100 Hispanic-Americans. It is estimated that 2.5 million Americans are heterozygous carriers for the sickle-cell trait.
France
As a result of population growth in African-Caribbean regions of overseas France and immigration from North and sub-Saharan Africa to mainland France, sickle-cell disease has become a major health problem in France. SCD has become the most common genetic disease in the country, with an overall birth prevalence of 1/2,415 in mainland France, ahead of phenylketonuria (1/10,862), congenital hypothyroidism (1/3,132), congenital adrenal hyperplasia (1/19,008) and cystic fibrosis (1/5,014) for the same reference period. In 2010, 31.5% of all newborns in mainland France (253,466 out of 805,958) were screened for SCD (this percentage was 19% in 2000). 341 newborns with SCD and 8,744 heterozygous carriers were found representing 1.1% of all newborns in mainland France. The Paris metropolitan district (Île-de-France) is the region that accounts for the largest number of newborns screened for SCD (60% in 2010). The second largest number of at-risk is in Provence-Alpes-Côte d'Azur at nearly 43.2% and the lowest number is in Brittany at 5.5%.
United Kingdom
In the United Kingdom (UK), it is thought that between 12,000 to 15,000 people have sickle cell disease with an estimate of 250,000 carriers of the condition in England alone. As the number of carriers is only estimated, all newborn babies in the UK receive a routine blood test to screen for the condition. Due to many adults in high risk groups not knowing whether they are carriers, pregnant women prior to birth and both partners in a couple are offered screening in order to obtain counseling if they are discovered to have sickle cell trait. In addition blood donors from those in high risk groups are also screened to confirm whether they are carriers and whether their blood filters properly. Donors who are found to be carriers are then informed and their blood, while often used for those of the same ethnic group, is not used for those with sickle cell disease who require a blood transfusion.
Middle East
In Saudi Arabia about 4.2% of population carries the sickle-cell trait and 0.26% has sickle-cell disease. The highest prevalence is in the Eastern province where approximately 17% of the population carries the gene and 1.2% has sickle-cell disease. In 2005 in Saudi Arabia a mandatory pre-marital test including HB electrophoresis was launched and aimed to decrease the incidence of SCD and thalassemia.
India
Sickle-cell disease is common in the tribal people (due to keen relations) of central India who share a genetic linkage with the African race, where the prevalence has ranged from 9.4 to 22.2% in endemic areas of Madhya Pradesh, Rajasthan, Chhattisgarh and Odisha.
Caribbean Islands
In Jamaica, 10% of the population carries the sickle-cell gene, making it the most prevalent genetic disorder in the country.

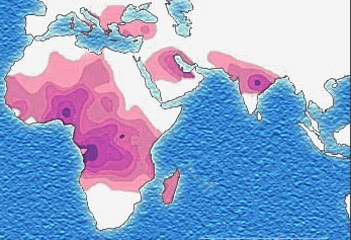
Distribution of the sickle-cell trait shown in pink and purple
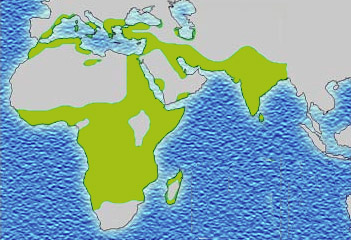
Historical distribution of malaria (no longer endemic in Europe) shown in green